Anestésico locales
1. INTRODUCCIÓN
La utilización de anestésicos locales (AL) constituye un pilar fundamental en el arsenal terapéutico actual del anestesiólogo, tanto para la realización de técnicas anestésicas, como para analgesia postoperatoria. Su desarrollo ha ido paralelo al descubrimiento e introducción en la clínica de sustancias capaces de “producir anestesia”. Estos fármacos, cuando se utilizan a concentraciones adecuadas, impiden la generación, propagación y conducción de impulsos nerviosos de manera reversible y temporal, de forma tal que producen supresión de la sensibilidad de una determinada zona (analgesia/anestesia) evitando la necesidad de la pérdida de consciencia, como sucede cuando aplicamos una anestesia general.
La historia de los anestésicos locales se remonta a 1855 (Fig. 1), cuando Galdeck aisló la cocaína en las hojas de la coca, pero su introducción en la clínica no se produce hasta el año 1884 por Koller(1), quien la aplica en una anestesia ocular para la corrección quirúrgica de un caso de glaucoma. Posteriormente Halstead la utilizó para el bloqueo de troncos nerviosos y August Bier por vía espinal en 1899.
Las moléculas benzocaína, procaína y tetracaína se introducen como AL en los primeros años del siglo XX, y no es hasta 1944 cuando comienza a usarse las moléculas tipo amida (lidocaína).

Figura 1
Historia de los anestésicos locales
2. MECANISMO DE ACCIÓN
Los AL son moléculas que bloquean la transmisión nerviosa (producción y propagación del potencial de acción) merced a su capacidad de bloquear de forma reversible los canales de Na+ voltaje-dependientes de la membrana de la célula nerviosa. Para comprender este mecanismo de acción es fundamental conocer los aspectos básicos de la fisiología y anatomía funcional de las estructuras nerviosas.
Algunos axones o fibras de los nervios periféricos se encuentran envueltos por una gruesa cubierta de mielina que las aísla de la fibra. Esta envoltura presenta interrupciones periódicas (denominadas nódulos de Ranvier), lo que confiere una gran rapidez a la hora de transmitir información, ya que los potenciales de acción “viajan” de un nódulo de Ranvier al siguiente. A este fenómeno se le denomina “conducción saltatoria”, y es la forma habitual de propagarse el potencial de acción en las fibras nerviosas. Sin embargo, las fibras nociceptivas y eferentes posganglionares son delgadas y carecen de vaina de mielina, por lo que la conducción del potencial de acción es continua y más lenta.
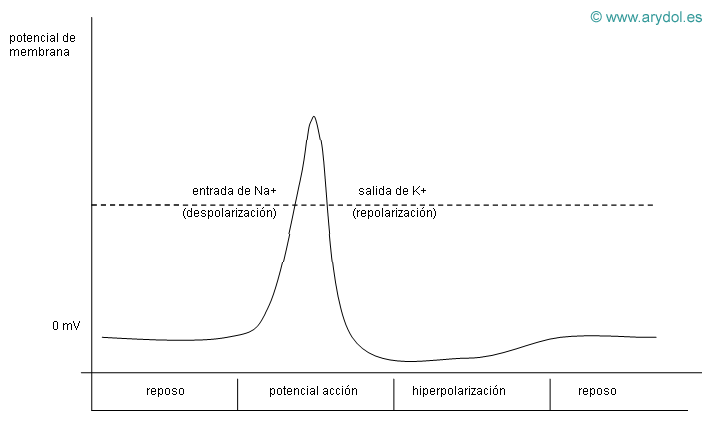
En condiciones normales, la membrana de la célula nerviosa se compone de canales iónicos que son relativamente impermeables a los iones Na+. Además, existen mecanismos activos de expulsión de Na+ al medio extracelular y de entrada de K+ (ATPasa Na+/K+). Esta situación se traduce en unas concentraciones iónicas muy diferentes a ambos lados de la membrana, lo que genera una diferencia de potencial que, en situación de reposo, es de unos – 65 mV intracelular. Un potencial negativo significa que existe un exceso de cargas negativas en la cara interna de la membrana celular.
Cuando un estímulo eléctrico, mecánico o químico actúa sobre la membrana y sobrepasa el umbral de excitación de la misma, se genera una despolarización de la membrana que se propaga por la fibra nerviosa, a este fenómeno se le denomina potencial de acción (Fig. 2).
Los cambios en el voltaje de la membrana provocan cambios conformacionales en los canales iónicos, que pasan a permitir un flujo iónico a su través (aumentan conductancia). Este incremento de la conductancia para el Na+ tendrá lugar en sentido de entrada a la célula. El potasio, por el contrario saldrá al medio extracelular. El flujo de Na+ es comparativamente mayor, por lo que la despolarización generará una entrada neta de cargas positivas, con la consiguiente elevación del potencial de la membrana nerviosa, hasta unos +35 mV.
Potencial de acción de la célula nerviosa
El aumento de permeabilidad al Na+ es muy rápido, pero fugaz, ya que la propia despolarización induce un cambio en el canal iónico del Na+, haciéndolo impermeable y disminuyendo por tanto su conductancia (fase de inactivación). Posteriormente se produce la fase de repolarización por aumento de la conductancia al K+, que sale de la célula hasta que el número de iones que salen iguala al de iones de Na+ que habían entrado, restituyéndose así el potencial de reposo de la membrana.
Todo este proceso transcurre en milisegundos, por lo que una fibra puede transmitir varios cientos de impulsos en un segundo.
Los AL impiden la generación y propagación de estos potenciales de acción. Para ello es preciso que la molécula alcance su lugar de acción (canal de Na+). Este canal proteico se encuentra en la cara citoplasmática de la membrana celular, por lo que el AL debe atravesar las envolturas y membranas de la célula (epineuro, perineuro y endoneuro) y, una vez en el medio intracelular, interaccionar con el canal iónico.
Un mecanismo de acción secundario de los AL es el cierre del canal iónico por compresión provocado por la expansión de la membrana celular al introducirse el AL en la misma.
Existen varios tipos de fibras nerviosas (Fig.3), ya que las diferencias entre el tipo y grosor de sus envolturas les confieren distintas propiedades bioeléctricas y también determinan su comportamiento cuando son expuestas a un AL.
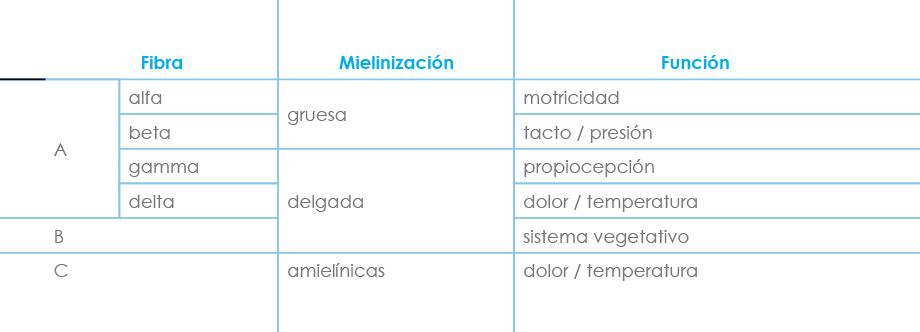
Figura 3
Clasificación funcional de las fibras nerviosas
El bloqueo de las fibras delgadas y amielínicas (fibras C, B y A delta, responsables de la inervación simpática y de la transmisión térmica y del dolor) es más rápido y prolongado, ya que precisan menor cantidad de AL para ser bloqueadas. Esta es la razón por la que el uso de soluciones anestésicas con escasa concentración puede permitir una buena analgesia sin repercusión significativa sobre la función motora (bloqueo diferencial).
La cronología del bloqueo nervioso sigue el siguiente orden:
- Bloqueo simpático con vasodilatación periférica y aumento de la temperatura cutánea (fibras B).
- Pérdida de la sensibilidad dolorosa y térmica (fibras A delta y C)
- Pérdida de la propiocepción (fibras A gamma)
- Ausencia de la sensibilidad al tacto y a la presión (fibras A beta)
- Parálisis motora (fibras A alfa)
Otro concepto interesante es el de concentración mínima inhibitoria (CMI), que se refiere a la concentración mínima de AL necesaria para bloquear un determinado tipo de fibra nerviosa, siendo diferente para cada AL, lo que nos permite clasificarlos según su potencia. Asimismo, existen distintos subtipos de canales de Na+ en las fibras nerviosas (tetrodotoxinsensibles y tetrodotoxin –resistentes). La progresiva caracterización de estos canales está ampliando nuestro conocimiento acerca de los mecanismos de acción de los AL. Ahora sabemos que ciertos AL muestran mayor afinidad por unos subtipos determinados, lo que explica parte de su perfil clínico(4).
Otros aspectos fundamentales que debemos tener en cuenta de los AL son la ionización, la potencia, la liposolubilidad, el tiempo de latencia, la duración de acción,... Todos los AL comparten un mismo mecanismo de acción, pero las diferencias que presentan en lo que respecta a estas características son las que nos permiten elegir el más adecuado y usarlo juiciosamente en función de cada situación clínica.
3. ESTRUCTURA QUÍMICA
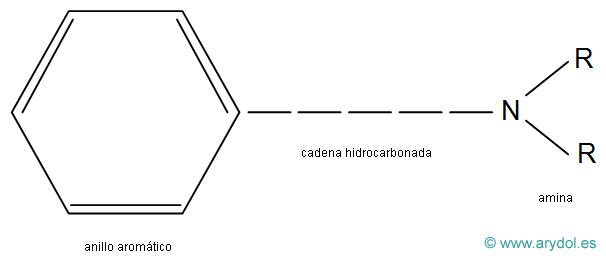
Todos los AL comparten una estructura química básica común (Fig. 4), compuesta por:
- una amina secundaria o terciaria (base aceptora de protones y, por lo tanto, capaz de cargarse positivamente), condiciona la hidrofilia de la molécula.
- un anillo aromático (núcleo bencénico), confiere liposolubilidad.
- una cadena intermedia hidrocarbonada que une la amina básica con el anillo aromático y es variable en longitud y ramificaciones. Presenta un enlace tipo aminoéster (CO) o aminoamida (CNH).
Estructura química básica común
En función de esta unión entre el núcleo aromático y la cadena hidrocarbonada, los AL se clasifican en ésteres y amidas (Fig.5).

Figura 5
Clasificación de los anestésicos locales
Las características farmacocinéticas y farmacodinámicas de los AL dependen en buena parte de su estructura química.
El anillo aromático condiciona las propiedades lipofílicas de la molécula y la amina secundaria o terciaria le proporciona carácter hidrofílico. Los AL son, por tanto, sustancias anfipáticas (la misma molécula presenta regiones hidrofílicas e hidrofóbicas).
El alargamiento y ramificación de la cadena hidrocarbonada aumenta la potencia analgésica. La presencia en esta cadena intermedia de un grupo éster o amida determina el metabolismo de la molécula. Los AL con unión aminoéster son fácilmente hidrolizados en el plasma por las colinesterasas y, en cambio, los que tienen una unión aminoamida son metabolizados por los microsomas hepáticos. El uso clínico actual de los AL se ha dirigido hacia el desarrollo de las aminoamidas, debido a su mayor estabilidad y menor potencial alergénico.
POTENCIA
La liposolubilidad condiciona la potencia del AL, ya que los AL lipofílicos cruzan más fácilmente las envoluturas nerviosas. Los AL de mayor peso molecular y más lipofílicos suelen ser más potentes, más tóxicos y su efecto es más prolongado. El coeficiente de partición es una medida de la liposolubilidad de la molécula.
DURACIÓN DE ACCIÓN
La duración del efecto depende fundamentalmente de la liposolubilidad, la afinidad a las proteínas plasmáticas y el metabolismo del AL. A mayor liposolubilidad, mayor unión a proteínas plasmáticas y menor velocidad de difusión al torrente sanguíneo y de metabolización, por tanto mayor duración del efecto.
Asimismo, existen preparaciones comerciales con vasoconstrictor (generalmente adrenalina 1:200000, o lo que es lo mismo, 5 mcg/ml). La adición de adrenalina produce una vasoconstricción local que condiciona una absorción más lenta, menor pico plasmático (menor toxicidad) y prolonga la duración de acción. Estas preparaciones deben evitarse en zonas con flujo sanguíneo colateral deficiente (dedos, pene, pabellón auricular,…), en técnicas regionales intravenosas y en pacientes con cardiopatía isquémica, arritmias previas o tratados con inhibidores de la monoaminooxidasa (IMAO) o antidepresivos triciclicos.
LATENCIA
El pKa determina la velocidad de inicio del bloqueo (latencia).
Los AL son bases débiles y pueden cambiar de estado al ionizarse aceptando protones.
El pKa es el pH al que el 50% de las moléculas se encuentran en forma ionizada y el otro 50% en forma no ionizada.
El pKa describe, por tanto, la proporción entre moléculas ionizadas (cationes) y no ionizadas (bases) presentes en una solución de AL.
Según la ecuación de Henderson-Hasselbach, pKa = pH + log ([cationes]/[bases])
El comportamiento de ambas formas (ionizada y no ionizada) es distinto. Las formas no ionizadas son más liposolubles y atraviesan con facilidad las membranas biológicas (envolturas nerviosas), lo cual es necesario para el inicio de acción del AL. Las moléculas en forma ionizada son más hidrosolubles, por lo que pueden pasar al interior celular y ejercer su acción a nivel de los canales de Na+ voltaje-dependendientes.
Los agentes con un pKa bajo (más cercano al pH fisiológico) tienen un inicio de acción más rápido, ya que al haber más formas no ionizadas disponibles, y por tanto ser mas liposoluble éstas atraviesan más rápidamente las membranas biológicas.
En las preparaciones comerciales el AL se encuentra en forma de sal de clorhidrato hidrosoluble y presenta un pH bajo, para que predominen las formas ionizadas y se asegure la estabilidad en solución acuosa. Una forma de intentar acelerar el inicio de acción del AL consiste en la alcalinización de la solución anestésica, ya que así aumenta la proporción de moléculas no ionizadas. Un exceso de alcalinización disminuye la estabilidad galénica de la disolución, con riesgo de precipitación del anestésico. Las dosis de bicarbonato sódico recomendadas para la alcalinización del AL son de 1 mEq /10mL de lidocaína o mepivacaína; y de 0,1 mEq/ 10mL de bupivacaína. Estos beneficios teóricos no están claramente avalados por los estudios in vivo realizados(5). La carbonatación da lugar a un aumento de la difusión del AL a través de la vaina nerviosa, produciendo un comienzo de acción más rápido y una disminución en la concentración mínima necesaria para el bloqueo de conducción, debido a que la difusión del anhídrido carbónico a través de la membrana nerviosa disminuye el pH axoplásmico.
El calentamiento del AL antes de su administración a 37ºC disminuye el pKa, aumentando la cantidad de fármaco no ionizado, por lo que disminuye el período de latencia y mejora la calidad del bloqueo. Otro efecto clínico significativo del calentamiento de la solución de AL consiste en una disminución de su viscosidad, lo que condiciona una mayor difusión cuando se administra por vía subaracnoidea, alcanzándose un nivel de bloqueo más extenso(6).
Otra posibilidad para acortar la latencia del AL consiste en la adición de hialuronidasa, que pretende acelerar la difusión del AL en el tejido.
Otro aspecto relacionado con el pH que debemos tener en cuenta es el fenómeno de taquifilaxia o tolerancia (disminución del efecto de un AL con inyecciones repetidas) que parece deberse a una acidificación progresiva en el lugar de inyección, siendo más frecuente con los AL de pKa menor.
ESTEREOISOMERÍA
Los AL son sustancias que presentan un carbono asimétrico (aquél unido a cuatro radicales distintos entre sí). Esta particularidad les permite aparecer en dos formas tridimensionalmente distintas, imágenes especulares la una de la otra, denominadas enantiómeros. La mezcla equimolecular de ambos enantiómeros se denomina mezcla racémica.
Cada una de estas dos formas presenta un comportamiento óptico característico. Cuando el AL es expuesto a la luz polarizada, se produce un giro en ésta última. La molécula que produce una rotación de la luz en sentido horario, se denomina enantiómero S (levo, L, ), mientras que el enantiómero R (dextro, D, +) produce una rotación de la luz polarizada en sentido antihorario.
La tecnología actual permite separar ambos enantiómeros a un coste razonable, lo que ha dado lugar a la aparición de soluciones anestésicas compuestas por un enantiómero puro concreto. Esta posibilidad se acompaña de los beneficios clínicos derivados de los enantiómeros S, menos tóxicos, más potentes y con una acción más prolongada que la mezcla racémica del AL. Son enantiómeros S puros la ropivacaína y la levobupivacaína. Aunque las propiedades farmacodinámicas de ambos enantiómeros deberían ser las mismas, los estudios demuestran una mayor potencia de la levobupivacaína con respecto a la mezcla racémica de bupivacaína. Esto no se debe a diferencias en la liposolubilidad de las moléculas, que es la misma, sino que parece relacionarse con la interacción con una proteína, que condiciona una mayor conductancia para el K+(7).

Figura 6
Propiedades farmacocinéticas-farmacodinámicas de los AL más empleados
4. TOXICIDAD
Las reacciones adversas relacionadas con los AL pueden ser clasificadas:
RELACIONADAS CON SU MECANISMO DE ACCIÓN
Hipotensión arterial en relación con el bloqueo simpático, bloqueo no deseado (anestesia espinal total, parálisis frenica tras bloqueo del plexo braquial,…).
RELACIONADAS CON SU METABOLISMO
Metahemoglobinemia en relación con ortotoluidina (metabolito de la prilocaína), convulsiones por xilidida (metabolito de la lidocaína).
REACCIONES ALÉRGICAS(8)(9)
Las reacciones anafilácticas se han relacionado casi exclusivamente con el ácido paraaminobenzoico (PABA), metabolito de los ésteres (de escasa utilización clínica, salvo tal vez las soluciones de uso tópico con cocaína en cirugía ORL). Se considera que las reacciones alérgicas a AL tipo amida son excepcionales, si bien se han observado reacciones alérgicas locales (dermatitis) de tipo hipersensibilidad retardada tipo IV en relación con lidocaína. Los responsables de reacciones alérgicas pueden ser los conservantes (metilparabén) o los antioxidantes (bisulfitos), presentes en las preparaciones comerciales de AL.
TOXICIDAD TISULAR LOCAL
Fundamentalmente a nivel del tejido muscular y nervioso cuando usamos concentraciones elevadas.
Un caso particular de toxicidad local a nivel del sistema nervioso, lo constituyen los Síntomas Neurológicos Transitorios (TNS). La clínica consiste en la aparición de dolor en las primeras 24 horas tras la realización de una anestesia subaracnoidea, que afecta a la región glútea y/o extremidades inferiores, sin otros datos en la exploración física, radiológica o electrofisiológica. El dolor desaparece en un máximo de 5 días, sin dejar secuelas. En los últimos años se han realizado metaanálisis que muestran una asociación entre este síndrome y la lidocaína de forma más consistente que con el resto de AL(10).
TOXICIDAD SISTÉMICA
En relación con altas concentraciones de AL en el torrente sanguíneo, bien por inyección intravascular directa o por difusión al sistema vascular.
Los órganos diana de la toxicidad sistémica de los AL son fundamentalmente el sistema nervioso central y el cardiovascular, siendo las dosis tóxicas para el primero inferiores a las del segundo. Estos efectos adversos vienen derivados del bloqueo de canales de Na+ en estos órganos diana, si bien existen otros mecanismos, como la interferencia con la respiración celular a nivel de las mitocondrias(11).
Generalmente son más precoces los síntomas y signos derivados del SNC, precediendo a las alteraciones cardíacas y hemodinámicas. Con frecuencia esta toxicidad sistémica se acompaña de histéresis (la clínica comienza con una concentración plasmática determinada y se mantiene incluso tiempo después de que dicha concentración haya disminuido hasta niveles no considerados tóxicos). La clínica habitual es la siguiente:
SNC: inicialmente aparecen inquietud, sabor metálico con entumecimiento oral y lingual, acúfenos, disartria, temblor,…; posteriormente puede aparecer actividad convulsiva y, finalmente, coma y parada respiratoria.
Cardiovascular: pueden causar inicialmente hipertensión y taquicardia para, posteriormente, aparecer hipotensión, bradicardia, bloqueo AV y, finalmente, arritmias malignas o parada cardíaca.
Incluso cuando no se realiza una punción intravascular, una fracción del AL inyectado pasa por difusión al torrente circulatorio. La proporción de AL que accede a la sangre del individuo será la responsable de la toxicidad sistémica y su cuantía dependerá fundamentalmente de la vascularización del lugar de inyección.
Podemos ordenar de mayor a menor difusión vascular: intravenoso > traqueal > interpleural > intercostal > caudal > paracervical > epidural > braquial > ciático > subcutáneo > subaracnoideo
La inyección fraccionada, la aspiración frecuente, la adición de vasoconstrictor y el respeto de las dosis máximas de cada AL (Fig. 7), son buenas prácticas encaminadas a disminuir la cantidad de anestésico local que accede al interior de los vasos.
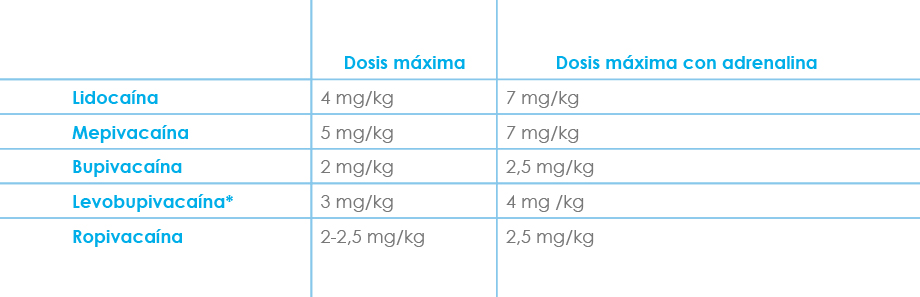
Figura 7
Dosis máximas recomendadas en la práctica clínica
*Teóricamente las dosis máximas son las mismas que con bupivacaína, pero debido a su menor toxicidad podrían emplearse dosis mayores. Con las dosis expresadas en la tabla los autores no observaron ningún efecto tóxico cuando se administra en bloqueo nervioso periférico.
La concentración plasmática es dosis dependiente y para la mayoría de AL existe una relación lineal entre dosis y concentración. Sin embargo, determinadas condiciones favorecen la toxicidad (niveles tóxicos sanguíneos por sobredosificación o inyección intravascular inadvertida, patologías asociadas, edades extremas, eventos de hipoxia o acidosis,…), pero también hay situaciones en las cuales el AL puede ser absorbido con demasiada rapidez, como cuando administramos los AL en bolo o en infusión continua en la herida quirúrgica.
Debemos recordar que la adición de un vasoconstrictor a la solución de AL, como la adrenalina 1:200000, disminuye el sangrado en la región infiltrada y reduce la absorción local del AL, aunque su efecto depende de la vascularización del lugar de inyección y de las propiedades vasoactivas del propio AL.
Además de la concentración que el AL alcanza en la circulación, existen multitud de factores que afectan a la toxicidad, de tal forma que una misma concentración plasmática puede ser tolerada por un paciente y letal para otro. En este sentido parecen tener efectos protectores la premedicación con una benzodiacepina (siempre que no acarree hipoventilación y acidosis) y la administración de oxígeno.
Un estudio de experimentación animal (12) demostró que la infusión intravenosa de AL en ovejas anestesiadas se correlacionaba con concentraciones plasmáticas mayores que la misma dosis en ovejas conscientes, debido a diferencias en la distribución del fármaco y su aclaramiento. Sin embargo, la mortalidad fue significativamente mayor en el grupo que no recibió anestesia general. Todo esto pone de manifiesto la necesidad de relativizar las dosis máximas asumibles con seguridad de AL, adaptándolas a cada situación clínica concreta. Por lo tanto, las dosis máximas no deben ser interpretadas en términos absolutos (13)(14).
El tratamiento de la intoxicación por AL consiste en la interrupción de la administración del anestésico, oxigenoterapia, corrección de acidosis, benzodiacepina / tiopental para tratar las convulsiones, reposición volumétrica, control de vía aérea y soporte inotrópico (adrenalina a dosis elevadas) si es necesario. En ocasiones se hace imperiosa la aplicación de un marcapasos. En caso de parada cardiorrespiratoria, se recomienda realizar RCP avanzada prolongada (>30 minutos). Se han comunicado casos de terapéutica satisfactoria con emulsión lipídica al 20%(15-17). Los efectos secundarios derivados de este tratamiento son tromboflebitis, inmunosupresión, embolia grasa y anafilaxia, entre otros.
Se considera que la toxicidad más temida es la derivada de la bupivacaína, por su cinética particular, que determina una unión a los receptores de Na+ cardíacos de forma más consistente. El fracaso en la reanimación de las paradas cardíacas asociadas a la utilización de bupivacaína en mujeres embarazadas dejó claro que los AL no eran tan inofensivos como se pensaba. Está documentado que la bupivacaína produce arritmias ventriculares malignas por acción directa sobre el corazón (efecto inotrópico negativo o por bloqueo de la conducción miocárdica) o bien por acción indirecta (bloqueo o desequilibrio de la inervación autonómica).
Existen multitud de estudios que pretenden comparar la toxicidad relativa de los distintos AL(18). Aberg(3) fue el primero en observar que el estereoisómero L de la mezcla racémica de bupivacaína es menos tóxico que estereoisómero D. Actualmente hay comunicados casos clínicos en los cuales la levobupivacaína produce alteraciones neurológicas tras inyección accidental endovenosa, pero en ningún caso hubo cardiotoxicidad. Bardsley y col. comprueban que tras la administración endovenosa en voluntarios sanos, levobupivacaína presenta menos toxicidad cardíaca que la bupivacaína racémica.
Algo parecido sucede con la ropivacaína. Los estudios realizados con dosis intravenosas de AL en voluntarios sanos para comparar la toxicidad aguda de ropivacaína frente a bupivacaína, demostraron que la ropivacaína es bien tolerada y presenta un potencial tóxico sobre el SNC y a nivel cardiovascular menor(19).
En resumen, mientras la bupivacaína ha dado lugar a paradas cardíacas irreversibles, los casos comunicados con levobupivacaína y ropivacaína pudieron ser reanimados satisfactoriamente(20)(21). Así se desprende de los informes de Ruetsch(22) del uso de ropivacaína en bloqueo ciático y de Pirotta de levobupivacaína en bloqueo del plexo braquial.
Los estudios clínicos que comparan efectos cardiovasculares y neurológicos inducidos por infusión endovenosa de levobupivacaína y ropivacaína en voluntarios sanos no encontraron diferencias(23). Ambos fármacos presentan una eficacia clínica semejante, sin embargo, la levobupivacaína es más potente y el perfil de seguridad de un fármaco debe valorarse comparando índices terapéuticos (margen entre dosis tóxica y terapéutica), por tanto, los resultados de los estudios de toxicidad deben considerarse junto a la potencia. Ambos anestésicos muestran perfiles de seguridad superponibles, sin embargo, debido a que la levobupivacaína es más potente que la ropivacaína, su margen de seguridad es más amplio.
Los autores consideramos que las dosis máximas en bolo expresadas en la tabla son orientativas. Pensamos que la mejor forma de evitar complicaciones secundarias a la utilización clínica de AL es seguir las normas de buena práctica clínica. Consideramos que el peso del paciente y el lugar donde administramos el fármaco (bloqueos centrales, periféricos, analgesia continua mediante infiltración de la herida quirúrgica) son factores fundamentales que debemos tener presentes. La monitorización clínica y el contacto verbal con el paciente cuando administramos el AL y en los momentos subsiguientes son cruciales para el diagnóstico precoz de la toxicidad. Con respecto a la infusión continua para analgesia postoperatoria, recomendamos utilizar ropivacaína o levobupivacaína, por presentar un mayor perfil de seguridad y facilidad para lograr un bloqueo diferencial.
5. LIDOCAÍNA
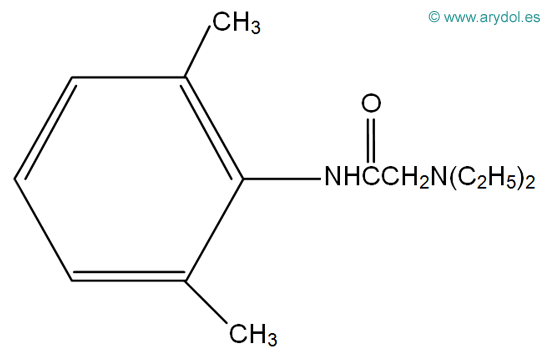
Se trata de un anestésico de corta duración de acción, con moderada unión a proteínas plasmáticas (56%). Existen presentaciones en ampollas (1% =10 mg/ml, 2%=20 mg/ml, 5%=50 mg/ml), como gel (2%) y en spray (10 %).
Usos clínicos: se utiliza tanto para bloqueos nerviosos periféricos, como para anestesiaanalgesia intradural y epidural. También se utiliza en anestesia regional intravenosa, para prevenir la reacción hemodinámica a las maniobras de intubaciónextubación, anestesia por infiltración y la forma de spray para anestesiar la vía aérea cuando realizamos intubación mediante fibrobroncoscopia.
Algunos anestesiólogos evitan su uso por vía intradural, dado su potencial neurotóxico(10). Se ha asociado con síntomas neurológicos transitorios, que se han comunicado más frecuentemente en caso de usarse lidocaína al 5% y a través de microcatéteres 32G para anestesia intradural.
6. MEPIVACAÍNA
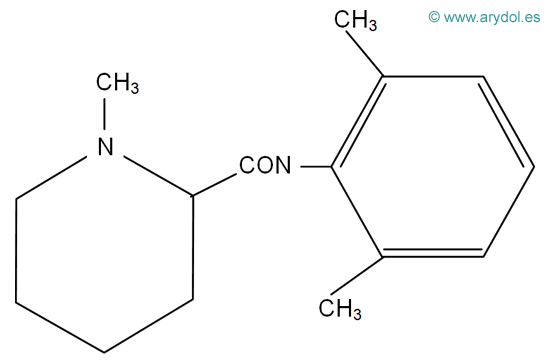
Los usos clínicos son similares a los de la lidocaína, sin embargo, su uso se ve limitado en obstetricia por su metabolismo prolongado en el feto y recién nacido.
Perfil anestésico similar a la lidocaína, aunque con una duración de acción algo mayor.
Presentaciones: ampollas 1% y 2% (10 o 20 mg/ml sin vasoconstrictor y con adrenalina 1:200000).
7. BUPIVACAÍNA
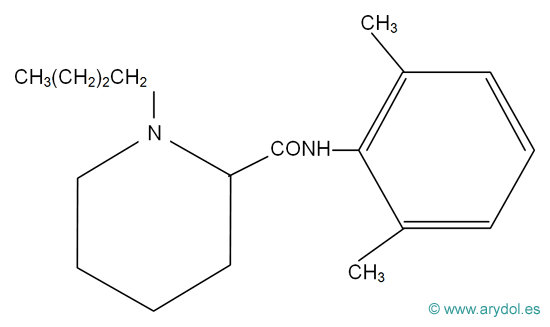
Anestésico tipo amida de duración de acción prolongada. Empleo generalizado en anestesia/analgesia de larga duración hasta el advenimiento de la levobupivacaína y la ropivacaína.
Existen preparaciones comerciales en ampollas a diferentes concentraciones: 0,25%, 0,5%, 0,755 sin vasoconstrictor. También al 0,25%y 0,5% con vasoconstrictor (adrenalina 1:200000) para retrasar el paso vascular del anestésico y, de eso modo, prolongar el efecto y disminuir su toxicidad.
También están disponibles soluciones hiperbáricas, que se depositan preferentemente en zonas declives cuando se usan por vía intradural. Esto permite que se pueda controlar el nivel del bloqueo variando la posición del paciente.
Con frecuencia se asocia a opioides para intensificar el bloqueo o controlar el dolor postoperatorio, aunque pueden aparecer efectos secundarios como prurito, náuseas, retención urinaria y, el más temido pero infrecuente, la depresión respiratoria.
8. LEVOBUPIVACAÍNA(24)(25)
Presentaciones: ampollas 0,25%, 0,5%, 0,75%. Bolsas para infusión continua a una concentración de 0,625 mg/ml y 1,25 mg/ml.
Se trata del enantiómero S puro de la bupivacaína. Anestésico tipo amida, con una elevada unión a proteínas plasmáticas (>97%). Presenta una potencia similar a la bupivacaína, pero con una duración algo mayor y una menor toxicidad cardiovascular y sobre el SNC.
A pesar su amplio margen terapéutico, en Europa no se recomiendan dosis mayores de 400 mg/día, 18.75 mg/hora epidural en adultos (12.5 mg/hora para uso obstétrico).
Usos clínicos:
- Epidural: concentraciones de 0,5-0,75% son adecuadas, con una duración del bloqueo sensorial de 69 horas. Para analgesia obstétrica pueden usarse concentraciones del 0,25% en bolo y, posteriormente, perfusión al 0,0625-0,125%. Para control de dolor postoperatorio emplearemos concentraciones entre 0,0625% y 0,125%.
- Intradural: concentración 0,5%. Dosis habitual de 15 mg. Duración del bloqueo motor de hasta 4 horas, con analgesia aún más prolongada. Comportamiento ligeramente hipobárico (1.0005 g/mL a 37 ºC).
- Bloqueo periférico: habitualmente a una concentración 0,5%, pero también puede utilizarse a mayor o menor concentración. Obtendremos una adecuada anestesia quirúrgica con una analgesia postoperatoria que se prolongará >16 horas. Asimismo puede utilizarse en perfusión continua a 0,0625% y 0,125% a nivel perineural.
- Debido a su perfil de seguridad, es de elección para administrar a través de infusión continua a través de catéteres colocados en la herida quirúrgica y para realizar una analgesia por infiltración prolongada.
- No se recomienda el uso de levobupivacaína en bloqueos paracervicales, en anestesia regional intravenosa, ni a elevadas concentraciones (0,75%) en obstetricia.
9. ROPIVACAÍNA
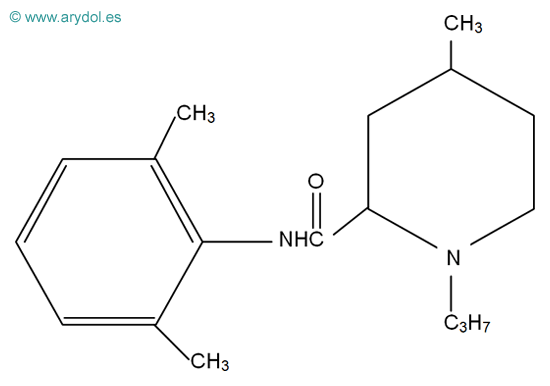
Presentaciones: ampollas 0,2%, 0,75%, 1%, bolsas de infusión 0,2%
Se trata de un enantiómero S puro, de duración de acción prolongada y escasa toxicidad.
Presenta una amplia unión a proteínas plasmáticas (94%), fundamentalmente a la α1glicoproteína ácida. Metabolismo hepático (como el resto de AL tipo amida).
Es menos potente que bupivacaína o levobupivacaína cuando se administra a dosis bajas. Sin embargo, este fenómeno no presenta apenas relevancia clínica, sobre todo cuando usamos altos volúmenes, como en el caso del bloqueo de troncos nerviosos periféricos.
Presenta un perfil adecuado para producir un bloqueo sensitivo diferencial cuando se usa a bajas concentraciones, con las ventajas que supone en las infusiones continuas y en procedimientos de cirugía ambulatoria.
La adición de un vasoconstrictor no parece acompañarse de beneficios clínicos.
En cuanto a la duración de acción, podemos decir que su uso a nivel epidural produce un bloqueo menos duradero que la bupivacaína, aunque este comportamiento no se reproduce con el uso a nivel periférico.
Una de las principales ventajas de la ropivacaína es su bajo perfil tóxico. La toxicidad cardíaca y nerviosa de la ropivacaína es menor que la de la bupivacaína. Además, la susceptibilidad a sus efectos tóxicos no está incrementada durante el embarazo y es bien tolerada por el feto, lo que, junto con el escaso bloqueo motor asociado, convierten a la ropivacaína en un atractivo agente para la analgesia epidural obstétrica y para la analgesia postoperatoria.
Usos clínicos:
- Intradural: por su potencial neurotóxico, la FDA no ha aprobado su uso por esta vía.
- Epidural: 7,5 mg/mL suelen se suficientes para producir anestesia quirúrgica. Se recomiendan 10 mg/ml si se requiere un bloqueo motor profundo. Ciertos procedimientos pueden realizarse satisfactoriamente con concentraciones de tan solo 3,75 mg/ml, con una rápida recuperación postanestésica. Una concentración de 2 mg/ml es adecuada para analgesia postoperatoria y, en el caso de analgesia para trabajo de parto, es suficiente con 1-2 mg/ml.
- Bloqueo de nervio periférico: es adecuada una concentración de 5 mg/ml.
- Infiltración local: proporciona analgesia eficaz y duradera tras la infiltración de la cicatriz quirúrgica tras, por ejemplo, colecistectomía(27), cirugía de mama(28) o reparación de hernia inguinal(29). La concentración adecuada será de 2,5 mg/ml. Los estudios arrojan buenos resultados con la infiltración tanto precomo post-incisional.
10. CREMA EMLA
La EMLA (Eutectic Mixture of Local Anesthetic) es una mezcla eutéctica (consigue disminuir su punto de fusiónrespecto a sus componentes de forma aislada). Consiste en una mezcla de prilocaína al 2,5% (punto de fusión 37ºC) y lidocaína al 2,5% (punto de fusión 67ºC), con un punto de fusión conjunto de 18ºC y rica en forma no ionizada para facilitar la penetración a través de piel y mucosas.
Se aplica sobre la piel (2,5 g / 10 cm2) y se cubre con un apósito oclusivo durante 1-2 horas. El efecto anestésico es máximo 30-60 minutos tras la retirada de la crema. En caso de piel lesionada o mucosas, el riesgo de toxicidad sistémica es mayor y el tiempo de oclusión efectivo y seguro es de 5-15 minutos.
Es útil para cirugía menor superficial y para punciones a través de piel o mucosas.
11. FUTURO
Actualmente, para proporcionar analgesia postoperatoria, recurrimos a la infusión de AL a través de un catéter. Una alternativa atractiva en fase de investigación y desarrollo consiste en cargar AL en un liposoma o matriz glucoproteica microcapsular. Los liposomas son microesferas de diferente e ínfimo tamaño, con una o varias bicapas lipídicas, separadas por compartimentos acuosos, lo que permite que durante su elaboración puedan incorporarse fármacos tanto liposolubles como hidrosolubles. Actualmente se está utilizando técnicas de administración liposomal para fármacos quimioterápicos y antifúngicos. Los AL también pueden cargarse en liposomas tanto en la fase acuosa como en la lipídica, para ser liberados en los tejidos tras su inyección. La liberación de los AL se ve influenciada por el tamaño de las vesículas, su estructura, el número de bicapas lipídicas presentes y la composición de las mismas.
Otras vías de investigación van dirigidas a encontrar moléculas con efecto AL más eficaces y seguras. La aplicación clínica de los enantiómeros S puros ya ha constituido un gran paso en este camino. Otras moléculas que están siendo testadas son la neosaxitoxina(30), bulleyaconitina(31),etc.
12. BIBLIOGRAFÍA
- Gunther B. Karl Koller: centennial of the discovery of local analgesia. Rev Med Chil 1984; 112 (11): 1181-85
- Ruetsch YA, Böni T, Borgeat A. From cocaine to ropivacaíne: the history of local anesthetic drugs. Curr Top Med Chem 2001; 1 (3): 175-82
- Aberg G. toxicological and local anesthetic effects of optically active isomers of two local anaesthetic compounds. Acta Pharmacol Toxicol 1972; 31 (4): 273-86
- Oda A, Ohashi H, Komori S. Characteristics of ropivacaine block of Na+ channels in rat dorsal root ganglion neurons. Anaesth Analg 2000; 91(5): 1213-20
- Chassard D, Berrada K, Boulétreau P. Alkalinization of local anesthetics: theoretically justified but clinically useless. Can J Anaesth 1996; 43(4): 384-93.
- Arai YC, Ueda W, Takimoto E, Manabe M. The influence of hiperbaric bupivacaine temperature on the spread of spinal anesthesia. Anesth Analg 2006; 102(1): 272-5
- Nau C, Vogel W, Hempelmann G. Stereoselectivity of bupivacaine in local anestheticsensitive ion channels of peripheral nerve. Anesthesiology 1999; 91(3): 786-95
- Gall H, Kaufmann R, Kalveram CM. Adverse reactions to local anesthetics: analysis of 197 cases. J Allergy Clin Immunol 1996; 97(4): 933-7
- Boren E, Teuber SS, Naguwa SM, Gershwin ME.A critical review of local anesthetic sensitivity. Clin Rev Allergy Immunol. 2007; 32(1):119-28
- Zaric D, Christiansen C, Pace NL Punjasawadwong Y. Transient neurologic symptoms after spinal anesthesia with lidocaine versus other local anesthetics: a systematic review of randomized, controlled trials. Anesth Analg 2005; 100(6): 1811-6
- Weinberg G. Local Anesthetic toxicity: different mechanism for different end points. Anesth Analg 2002; 94(2): 479-80
- Copeland SE, Ladd LA, Gu XQ, Mather LE. The effects of General Anestesia on whole body and regional pharmacokinetics of local anesthetics at toxic doses. Anesth Analg 2008; 106 (5): 1440-49
- Rosenberg PH, Veering BT, Urmey WF. Maximum recommended doses of local anesthetics: a multifactorial concept. Reg Anesth Pain Med 2004; 29(6): 564-75
- Heavener JE. Let´s abandon blanket maximum recommended doses of local ansthetics. Reg Anesth Pain Med 2004; 29(6): 524
- Brull SJ. Lipid emulsion for the treatment of local anesthetic toxicity: patient safety implications. Anesth Analg 2008: 106(5): 1337-9
- Warren JA, Thoma RB, Georgescu A, Shah SJ. Intravenous lipid infusion in the successful resuscitation of local anesthetic induced cardiovascular collapse after supraclavicular braquial plexus block. Anesth Analg 2008; 106(5): 1578-80
- Corman, Shelby L. Skledar, Susan J. Use of lipid emulsion to reverse local anestheticinduced toxicity. Annals Pharm 2007; 41(11): 1873-7
- Reiz S, Nath S. Cardiotoxicity of local anesthetic agents. Br J Anaesth 1986; 58(7): 736-46
- Scott DB, Lee A, Fagan D, Bowler GM, Bloomfield P, Lundh R. Acute toxicity of ropivacaine compared with that of bupivacaine. Anesth Analg 1989; 69(5): 563-9
- Abouleish EI, Elias M, Nelson C. Ropivacaine-induced seizure after extradural anaesthesia. Br J Anaesth 1998; 80(6): 834-4
- Müller M, Litz RJ, Hübler M, Albrecht DM. Grand mal convulsion and plasma concentrations after intravascular injection of ropivacaine for axillary braquial plexus blockade. Br J Anaesth 2001; 87(5): 784-7
- Ruetsch YA, Fattinger KE, Borgeat A. Ropivacaineinduced convulsions and severe cardiac dysrrithmia after sciatic block. Anesthesiology 1999; 90(6): 1784-6
- Stewart J, Kellett N, Castro D. The central nervous system and cardiovascular effects of levobupivacaine and ropivacaine in healthy vonlunteers. Anaesth Analg 2003; 97(2): 412-16
- McLeod GA, Burke D. Levopupivacaine. Anaesthesia 2001; 56(4): 331-41
- Foster RH, Markham A. Levobupivacaine. A review of its pharmacology and use as a local anaesthetic. Drugs 2000; 59(3): 551-79
- Simpson D, Curran MP, Oldfield V. Ropivacaine. A review of its use in regional anaesthesia and acute pain management. Drugs 2005; 65(18): 2675-17
- Bisgaard T, Klarskov B, Kristiansen VB. Multiregional local anaesthetic infiltration during laparoscopic cholecystectomy in patients receiving multimodal prophylactic analgesia: A randomised, doublebinded, placebocontrolled study. Anesth Analg 1999; 89(4): 1017-24
- Johansson A, Axelson J, Ingvar C, Luttropp HH, Lundberg J.. Preoperative ropivacaine infiltration in breast surgery. Acta Anaesthesiol Scand 2000; 44(9): 1093-8
- Mulroy MF, Burgess FW, Emanuelsson BM. Ropivacaine 0.25% and 0.5%, but not 0.125%, provides effective wound infiltration analgesia after outpatient hernia repair, but with sustained plasma drug levels. Reg Anesth Pain Med 1999; 24(2): 136-41
- Rodriguez-Navarro AJ, Lagos N, Lagos M, Braghetto I, Csendes A, Hamilton J et al. Neosaxitoxin as a local anesthetic: preliminary observations from a first human trial. Anesthesiology 2007;106(2): 339-45
- Wang CF, Gerner P, Wang SY, Wang GK. Bulleyaconitine A isolated from Aconitum plant displays longacting local anesthetic properties in vitro and in vivo. Anesthesiology 2007; 107(1): 82-90